NGS Services
Our Next Generation Sequencing (NGS) unit offer a wide range of services and our layout to provide the optimum sequencing strategy. We offer our NGS services in the six areas listed below:
Our Next Generation Sequencing (NGS) unit offer a wide range of services and our layout to provide the optimum sequencing strategy. We offer our NGS services in the six areas listed below:
High-throughput sequencing of whole genome (WGS) is a powerful technique that includes sequencing of all of an organis´s chromosomal DNA as well as DNA contained in the mitochondria or chloroplast. Using WGS not only can we can discover and confirm mutations (SNPs, INDEL) with better resolution, we can now comprehensively study chromosomal rearrangements, phenotype-causing sequence variant, etc.
Our service includes:
Sequencing Platform: NextSeq 550, Illumina
![]()
 The human exome contains about 180,000 exomes (~30 Mb) arrayed in about 22,000 genes and it represents less than 2% of the human genome, but contains ~85% of known disease-causing variants. The exome sequencing, is an alternative to whole-genome sequencing due the costs, efficiency and the easier interpretability of much lower data volume compared to whole genome sequencing. With exome sequencing, you can investigate the protein coding regions of the genome when sequencing an entire genome is not practical or necessary. The goal of this approach is to identify the functional variants across a wide range of applications, including population genetics, genetic disease and cancer studies.
The human exome contains about 180,000 exomes (~30 Mb) arrayed in about 22,000 genes and it represents less than 2% of the human genome, but contains ~85% of known disease-causing variants. The exome sequencing, is an alternative to whole-genome sequencing due the costs, efficiency and the easier interpretability of much lower data volume compared to whole genome sequencing. With exome sequencing, you can investigate the protein coding regions of the genome when sequencing an entire genome is not practical or necessary. The goal of this approach is to identify the functional variants across a wide range of applications, including population genetics, genetic disease and cancer studies.
Our service performs exome sequencing with NextSeq 550 (Illumina), that is a efficient and fast approach and offers a comprehensive exome sequencing solutions, from library preparation to data analysis. We offer different ways for exome capture that use transposons and optimized hybridization steps to simplify and shorten the protocol from several days to just a day and a half.
Our service includes:
Sequencing Platform: Illumina NextSeq 550.
![]()
High-throughput sequencing of RNA (RNA-seq) is a powerful suite of techniques to understand transcriptional regulation. Using RNA-seq, not only can we perform traditional differential gene expression analysis with better resolution, we can now comprehensively study alternative splicing, RNA editing, allele specific expression, and identify novel transcripts, both coding and non-coding RNAs.
Our service includes:
Sequencing Platform: NextSeq 550, Illumina
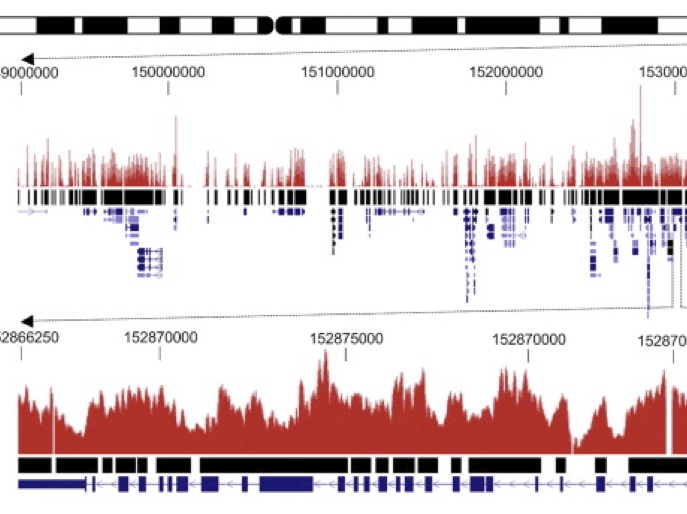 Sequence Capture of client-defined regions or fixed panels are an excellent way to detection of SNPs, InDels, and other genetic variants. This strategy helps to reduce the amount and cost of data required for characterization as compared to sequencing a whole genome. There are two basic approaches to re-sequence specific regions: Amplicon sequencing or in-solution sequence capture. In our service you can selectively enrich any genomic regions (customized gene panel) or order a fixed gene panels that have the greatest level of uniformity and allow researchers to identify disease-associated alleles within a population and others customer´s research needs. Benefits:
Sequence Capture of client-defined regions or fixed panels are an excellent way to detection of SNPs, InDels, and other genetic variants. This strategy helps to reduce the amount and cost of data required for characterization as compared to sequencing a whole genome. There are two basic approaches to re-sequence specific regions: Amplicon sequencing or in-solution sequence capture. In our service you can selectively enrich any genomic regions (customized gene panel) or order a fixed gene panels that have the greatest level of uniformity and allow researchers to identify disease-associated alleles within a population and others customer´s research needs. Benefits:
![]()
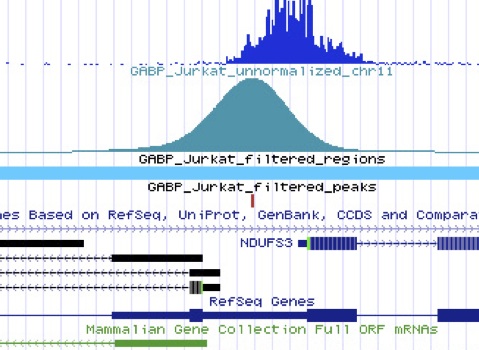 The combination of chromatin imunoprecipitación technique (chip) with the sequencing of the DNA fragments associated, allows information about the protein-DNA interactions along the entire genome and its regulation expressions to understand many biological processes and disease states. It determines whether specific proteins are associated with specific genomic regions, such as transcription factors on promoters or other DNA binding sites.
The combination of chromatin imunoprecipitación technique (chip) with the sequencing of the DNA fragments associated, allows information about the protein-DNA interactions along the entire genome and its regulation expressions to understand many biological processes and disease states. It determines whether specific proteins are associated with specific genomic regions, such as transcription factors on promoters or other DNA binding sites.
High-throughput sequencing on Illumina NextSeq 550 provides the ideal sequencing platform for ChIP experiments. Chip-seq library construct is done by Illumina TruSeq ChIP Sample Preparation Kits.
This service is only applicable in ChIP samples provided by our clients and allows us to:
Sequencing Platform: Illumina NextSeq 550.
![]()
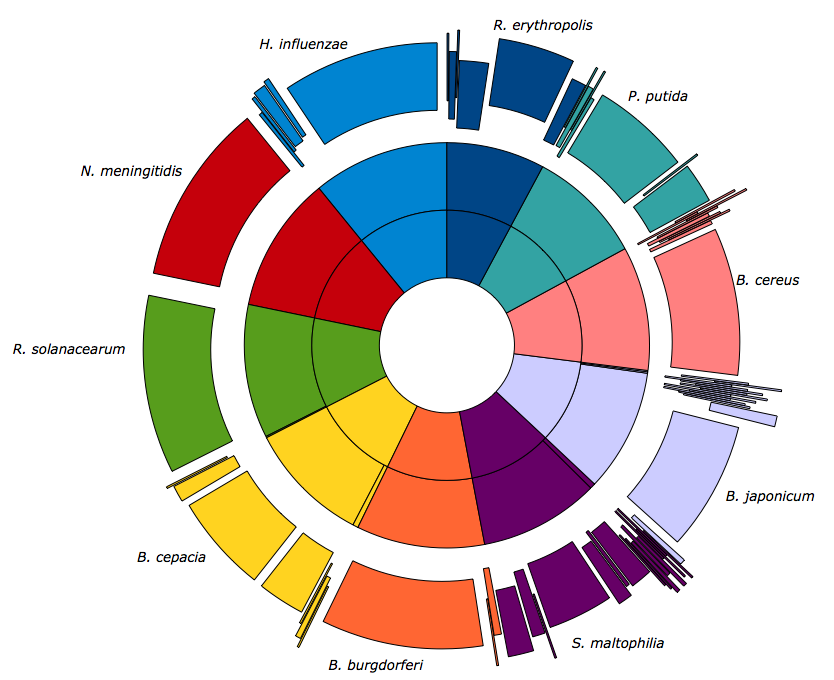 Metagenomics is the study of environmental samples where multiple microbial genomes are analyzed at the same time.The sequencing of whole genomes in a sample, allows to know what microorganisms are involved in certain diseases or conditions, or to characterize its function in different environments.These studies were performed using two approaches: 1-by shotgun sequencing of pools of microorganisms present in the sample and/or 2-The taxonomic identification by sequencing 16S or 18S rDNA region. In our service Metagenomic sequence reads are generated on Illumina platform taking advantage of the read lengths and deep sequencing capabilities.
Metagenomics is the study of environmental samples where multiple microbial genomes are analyzed at the same time.The sequencing of whole genomes in a sample, allows to know what microorganisms are involved in certain diseases or conditions, or to characterize its function in different environments.These studies were performed using two approaches: 1-by shotgun sequencing of pools of microorganisms present in the sample and/or 2-The taxonomic identification by sequencing 16S or 18S rDNA region. In our service Metagenomic sequence reads are generated on Illumina platform taking advantage of the read lengths and deep sequencing capabilities.
Applications:
Sequencing Platform: Illumina
![]()
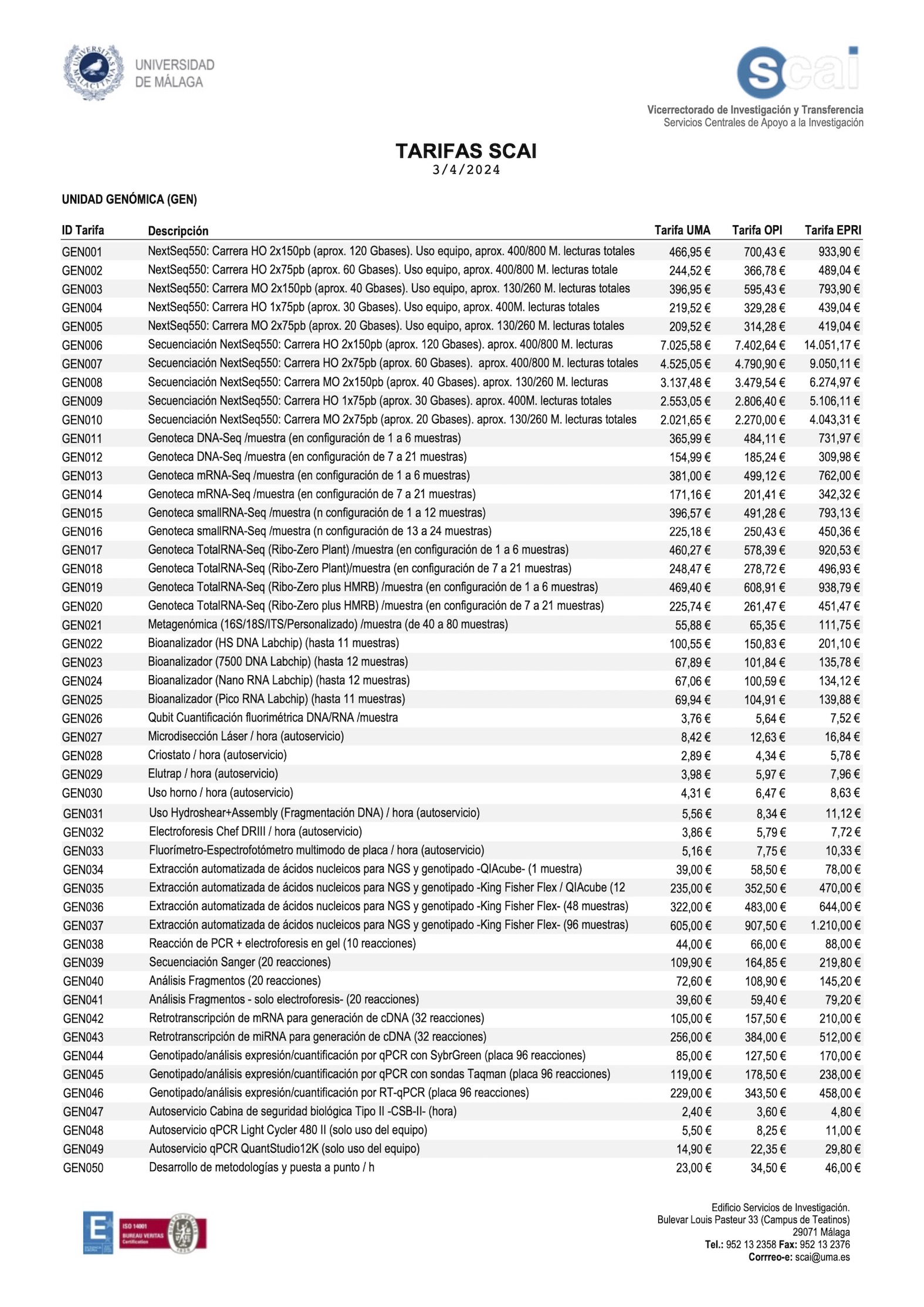
Puede descargar las tarifas pulsando aquí
*For other configurations of samples or batch, consult price according to technical valuation.
UMA: University of Malaga
OPI: Research Publics Organizations
PRI: Private Entities
Sample specification and submission guidelines
DNA Quality:
The DNA must be double stranded, without degradation and RNA contamination. DNA extraction by column purification method is strongly recommended (e.g. from Qiagen or any other supplier). Our service recommends avoiding phenol-chlroform in DNA extractions. The DNA can be resuspended in water or 10mM Tris-HCL, pH 7.5-8.5 (Eliminate the presence of EDTA in your samples).
The indicator of sample purity, OD260/280=1.8-2.0 and OD260/230≥1.8, are good indicators and when possible, our service recommends that DNA should be assessed by running a gel electroforesis.
The correct quantification of gDNA is essential in library construction. Our service recommends quantifying the starting genomic material by using a fluorescence based quantification method such a Qubit or picogreen assay. These measurement tend to be specific for double stranded DNA and are preferable to nanodrop or other UV based measurement. If your lab does not have a way of making a fluorescent based measurement, please measure with whatever method is available and indicate this information when submitting your sample.
RNA Quality:
For most RNA extraction, we recommend the RNeasy kit from Qiagen, however, you may use your own lab protocols or equivalent kits if they have yield high quality, without RNA
degradation. Use of degraded RNA can result in low yield, over-representation of the 5 ́ends or failure of the protocol. RNA that has DNA contamination will result in an underestimation of the amount of RNA used, for that reason, all RNA extraction must also be accompanied by DNase treatment. Qiagen protocols allow this step to be preformed on the column. Preventing RNase contamination is mandatory requirement when handling RNA samples. The purified RNA should be eluted or dissolved in RNAse-free water.
We recommend the use of Agilent Tecnologies 2100 Bioanalyzer to check the total RNA integrity. The specification for RNA Integrity Number (RIN) value of greater than 7 is requiered. Alternatively, if Bioanalyzer is not available, please run the RNA samples on a denaturing agarose gel to confirm the integrity of rRNA peaks.
The correct quantification of RNA is essential in library construction. Our service recommend quantifying the starting material by using a fluorescence based quantification method such a Qubit or Ribogreen assay. These measurement tend to be specific for RNA and are preferable to nanodrop or other UV based measurement. If your lab does not have a way of making a fluorescent based measurement, please measure with whatever available method and indicate this information when submitting your sample.
*Formalin-fixed, paraffin-embedded (FFPE) samples, that usually are strongly degraded, it is recommended to setup a pilot to confirm that materials are suitable.
*All samples undergo quality check (QC) before going into library preparation. Make sure to include enough volume to allow for 5 uL to be used in QC. The inputs listed below should remain after the completion of this QC. The following are minimum requirements for library construction. If your sample does not comply with this guidelines, please contact us and we can discuss your specific sample requirements.
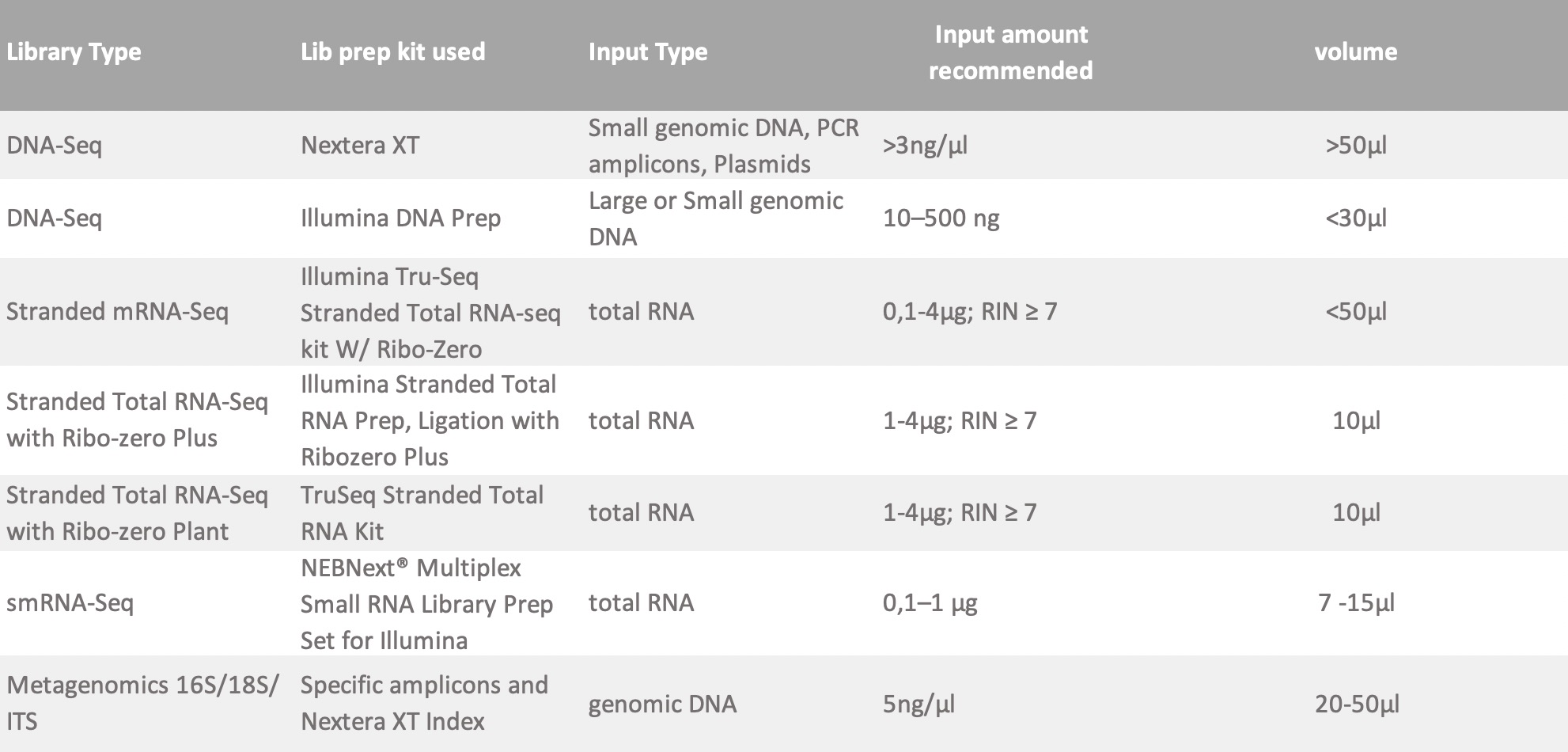
FOR PREPARED LIBRARY:
We do accept prepared libraries for sequencing. However, we do not guarantee the sequencing results for any customer prepared libraries.
Prepared libraries should be sent at ≥10 nM. A volume of 15ul at the minimum is required. Concentration should ideally be determined using fluorometric means (picogreen/Qubit). If you are pooling your libraries, the final pool should also be at at ≥10 nM with a volume of at least 15ul.
Submission Condition
TUBES:The samples should be placed in 1.5 ml microcentrifuge tubes sealed with parafilm, labelled and placed inside 50mL tubes. Make sure to write the sample number listed on your order sample sheet on the top the tube. To prevent sample tubes from moving during shipment, pack any remainning space with paper towel/tissue paper prior to sealing.
PLATES: Should you submit more than one sample, please use polypropylene PCR plates. Plates should be labeled with the order number and sample number range clearly marked, and should be sealed firmly with a foil plate seal capable of withstanding -80 C temperatures. Samples should be loaded on the plate in rows and not in columns.
*Transport should be in dry ice and send by Express company.
*Include a printed copy of the ordersheet with a description of the DNA/RNA extraction and/or enrichment protocol used. The description should incluye: Description of the sample: name, code, A260/280, concentration (ng/μl), specify quantitation methods (Pico/Ribogreen, nanodrop or/and spectrophotometre) a electrophoresis image of the EtBr-stained gel with the samples. Please include marker as a reference and clearly identify the contents and amount (in nanograms) of DNA of each lane. A Bioanalyzer profile can be incluye if it is available, mainly in RNA samples.
*Include a printed copy of your project quote in the package.
Sample Shipping to:
Laboratorio de Genómica y Ultrasecuenciación Universidad de Málaga
Edificio de Bioinnovación, 1a planta. puerta 104
Parque Tecnológico de Andalucía (PTA) C/Severo Ochoa, 34
29590 Campanillas-Málaga Spain.
Tlf. +34 951 952 780 (Attn. Josefa Gómez Maldonado)
Checklist for sample submission

All the parcel information must also be send to the contact service This email address is being protected from spambots. You need JavaScript enabled to view it.
Download guides: